A curious research on my genotype data from 23andMe

In the week that the first human gene-editing twins from China were born, I decided to share a story about what I’ve done to my genetic data to encourage people to take scientific findings with a grain of salt and to think critically about the results from years of biological researches. Coincidently, today we also past the 5-year anniversary where 23andMe first received FDA warning letter for providing diagnostic genetic test. As a device that is intended for diagnosis of disease, without solid premarket approval as the quality check to minimize safety issues, any false positive/negative information coming out from the product could lead to tragic life-changing decision-making.
Last month I went to an international annual conference held by American Society of Human Genetics, where I bought a discounted 23andMe test for ancestry + Health. As a second degree customer of 23andMe (all of my husband’s family members had their ancestry + health service done) and a Ph. D student who works in this field, having this service done is more like an entertainment rather than a mystery uncovering event. Two weeks ago I got my result, after confirming my ancestry is where I think I am from and a couple of “variants not detected” tabs from the health section. I downloaded my raw data and decided to do more.
Before I started doing anything, I realized there were already numerous services out there, some were free some were not, like impute.me, Promethease, DNA.land, etc. Despite how fancy these online tools are, they have one thing in common, which is carefree. But I want to have a little more responsibility by being a genetic researcher, I want to understand my genome by doing all the steps on my own.
Determined of my goal, first I want to know how much more information I can know beyond what was provided in 23andMe data. I want to ask “Can I confidently impute my genome to have a wider coverage? How much more variants could I impute confidently?”. Imputation uses comprehensive statistic modeling such as Markov Chain Monte Carlo (MCMC), which samples and computes expectation with respect to high dimensional probability distribution. Simply speaking, it uses available information to guess missing information using patterns that are shared between the two. My sample was genotyped on v5 chip (Illumina OmniExpress chip) which has ~650k variants , compared with 1000 genome 2015 version which characterized 88 million variants, there is still a lot of information in me that is left unknown.
The raw data is mapped to Genome Reference Consortium Human Build 37 (GRCh37), which is a synthetic hybrid that serves as an example but the sequence of which is not actually completely observed in any particular individual. With the latest human reference panel being GRCh38, GRCh37 is still widely used. Below is what the data looks like, it shows variant name, chromosome, position and genotype for a single variant in a row.
rsid chromosome position genotype
rs548049170 1 69869 TT
rs9283150 1 565508 AA
... ... ... ...
I did a count on my raw genotype, which returned me a table like this. Around 3% (18274 marked by ‘- -’) of my genotypes weren’t successfully done due to various reasons. I and D indicate insertion and deletion. Since humans are diploids, most of the time we have two copies of genetic information but the DNA in mitochondria only has 1 copy.
counts Genotype
18274 --
1256 A
109072 AA
9589 AC
39439 AG
319 AT
945 C
481 CG
39804 CT
10 D
1348 DD
47 DI
591 G
146998 GG
9614 GT
3 I
3440 II
1170 T
108594 TT
After removing genetic information from deletion/insertion, X chromosomes (not necessary but it requires special imputation), duplicated locations and mitochondria, I was left with 94% data (595249 variants). I chose to use the popular open source software SHAPEIT for haplotype inference and IMPUTE2 for the actual imputation. Both softwares are developed by University of Oxford and have well-documented manual for usage. So what is haplotype and why do we need to infer it? Haplotype is haploid genotype. Recall what we learned from biology class, during prophase I of meiosis, the homologues are paired, sections of chromosome from homologues are swapped (crossing over), therefore, we tend to inherit chunks of DNA information from one parent in a particular region. Since the recombination hotspots are relatively stable in a given population, if I can derive which chunks of my DNA tend to be inherited together in my genetic population (also called phasing), I could use other people’s genetic information whom are similar to mine to guess my missing genotypes!
I converted my filtered genotype data to PED and MAP format, and downloaded 1000 Genomes Phase 3 - for samples <10 reference panel is needed. Next thing is to figure out which strand the genotype was on, luckily, on the 23andMe website, it says all genotype information displayed are referred to positive (+) strand on GRCh37, which is the same strand used in 1000 Genome. After getting all the information I needed, I started haplotype inference, unfortunately, since I’m the only sample in the file, the program is still confused about strand issues since it hasn’t seen enough sample of to infer some of the homozygous alleles I have, like AA, CC, GG, TT. As a result, the program will drop those SNPs instead of continuing till the end. To solve this problem, I came up with a creative way to create a fake sample whose genotype is heterozygous on the position where my genotype is homozygous guided by the intermediate files generated by SHAPEIT. With this 4 steps process: a. check 1 sample PED file finding out the list of variants that was dropped due to ambiguous strand issue, b. create a drop list with variants that have other issues such as missing information or position duplication, c. append a fake sample to PED file that only has my genotype so that the program won’t be confused on positions where I am homozygous, d. run phasing algorithm to infer missing genotype. I used only used east Asian haplotype from 1000 Genomes, knowing this is where my ancestry is from and using it will be most accurate and time-saving. Finally, all the files are phased by chromosomes and ready for imputation. Things are pretty straightforward, the only headache part is to split chromosome by chunks and do imputation chunk by chunk. This takes some thinking because not all chromosomes are equal length (chr1 is the biggest and 21, 22 are the smallest) and not all variants are equally positioned, meaning a chuck boundary worked in one chromosomes doesn’t necessarily work in another. To solve that, I calculated how many chunks for each chromosomes from the beginning position to the end position and split the files with position evenly spaced tailored by individual chromosome.
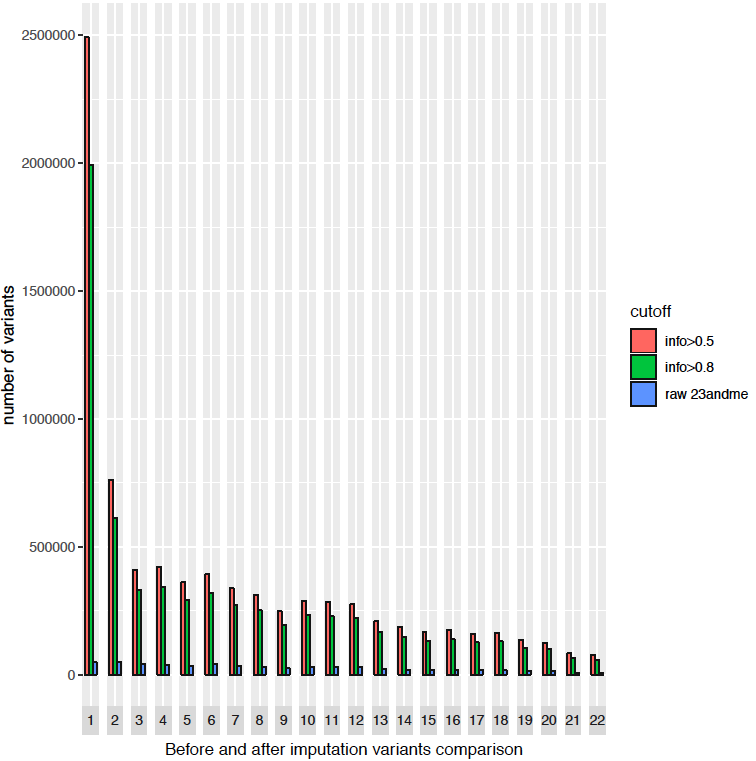
After doing all of the above, voilà! The imputation is all done! After concatenating the result into 1 file and filtering out the absolute low quality variants (info score<0.3), I got 8.6 million variants, which increased the information I got from 23andMe 15 times. I plotted the number of variants with different imputation quality cutoff by chromosome here, you can visualize how much information has been gained from imputation. Even with higher imputation quality threshold (info>0.8), there are still 6.5 million SNPs left. Of course, compared with 84 million known genetic polymorphism from 2015 Nature paper of 1000 Genomes project, this is just a fraction.
With more available information, what left undone is to look up SNPedia to find genetic risk with care.
The code of this project can be found on my github.